Built on Real Regulatory Workflows
Not generic templates. The platform mirrors how teams actually prepare medical device documentation — step-by-step, with structured tasks, organised folders, and clear "what to include" guidance.
Getting your medical device CE marked can feel complex. We break the process down into clear, simple steps — from a free MDR check to a full regulatory workflow, with built-in AI support.
Backed by


What People Are Saying
Breaking down regulatory compliance into bite-sized pieces.
Tell us what you're building and who it's for. We turn your input into a clear description that fits regulatory expectations.
We show you if MDR applies, what risk class you fall into, and the route you'll need to take.
Follow a structured plan to create your documentation step by step. Instead of starting from scratch, you get guidance, templates, checklists, and a clear sense of direction.
Start free. Upgrade when you're ready.
Trial
Free
no credit card required
No credit card · Cancel anytime
Pro
€299
per month · excl. VAT
Billed monthly · Cancel anytime
Pro Annual
€2,990
per year · excl. VAT · €249/mo
Billed annually · Cancel anytime
Shaped and vetted by regulatory specialists. Grounded in official EU guidance.
Used by multiple industry experts.
Not generic templates. The platform mirrors how teams actually prepare medical device documentation — step-by-step, with structured tasks, organised folders, and clear "what to include" guidance.
From day one, you work in a reviewable structure. Track progress, maintain consistency across documents, and reduce gaps that slow down internal reviews.
Know what's required — and why. Every step helps you understand the rationale behind decisions, so you can align stakeholders and defend choices with confidence.
Our partners




Plan, draft, and maintain your MDR documentation in one guided workflow.
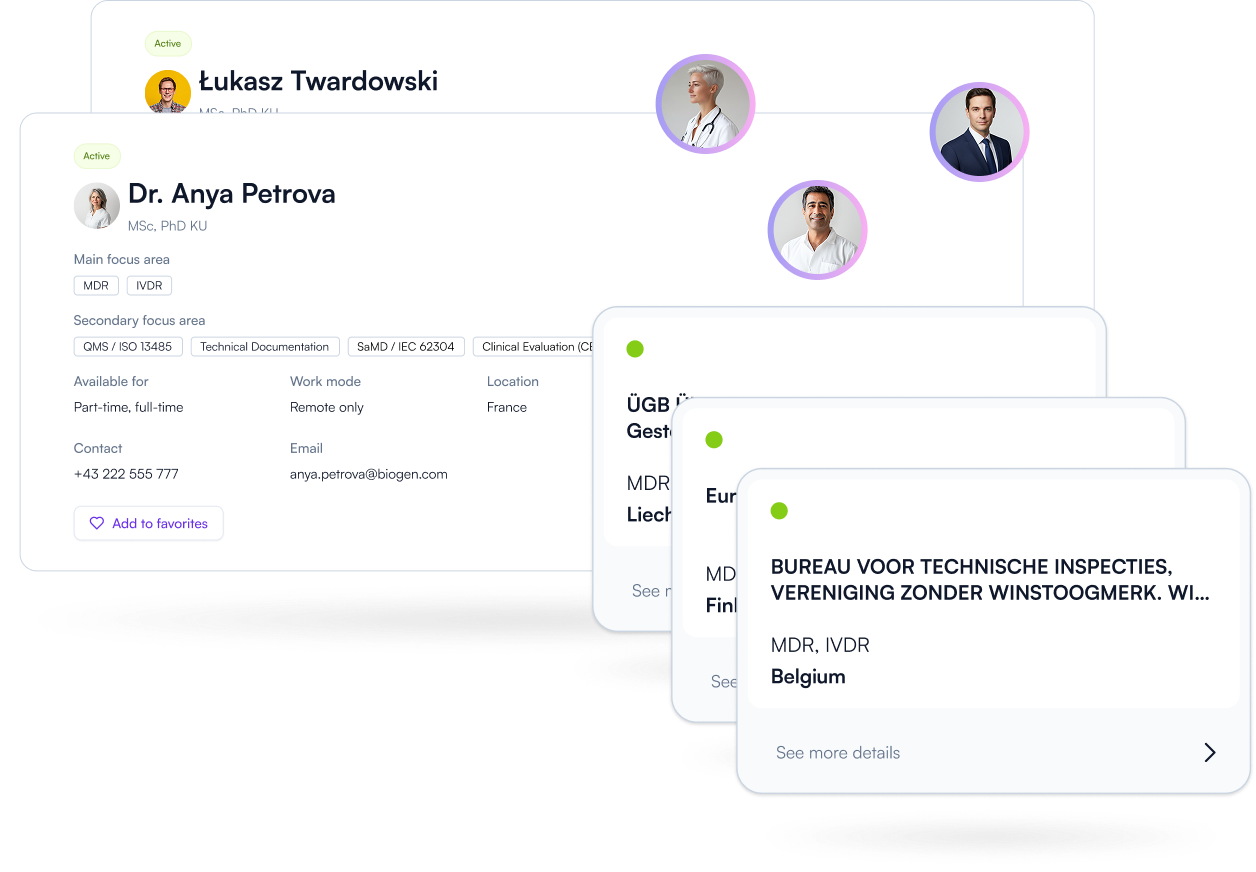
Find the right experts faster. Compare specialization, scope, and collaboration models — connect when your project needs regulatory support. Less searching, better fit, faster progress.
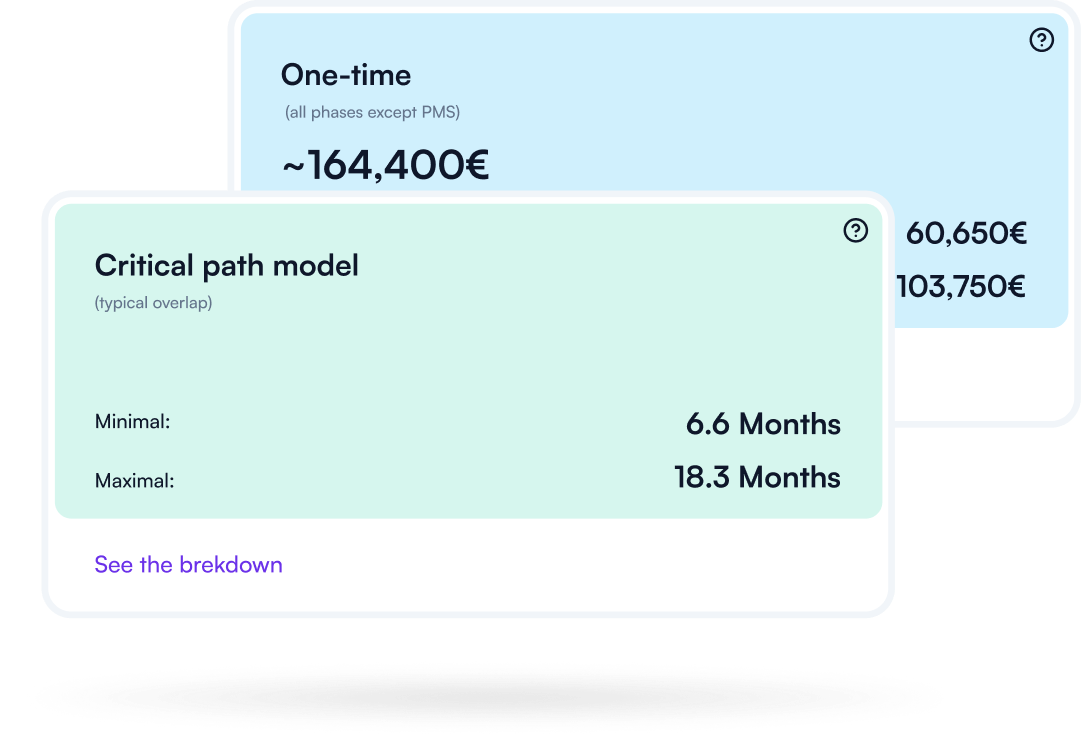
Understand what drives effort and cost across your compliance journey — so you can plan realistically, prioritize, and avoid late surprises. Make timelines and budgets defensible.
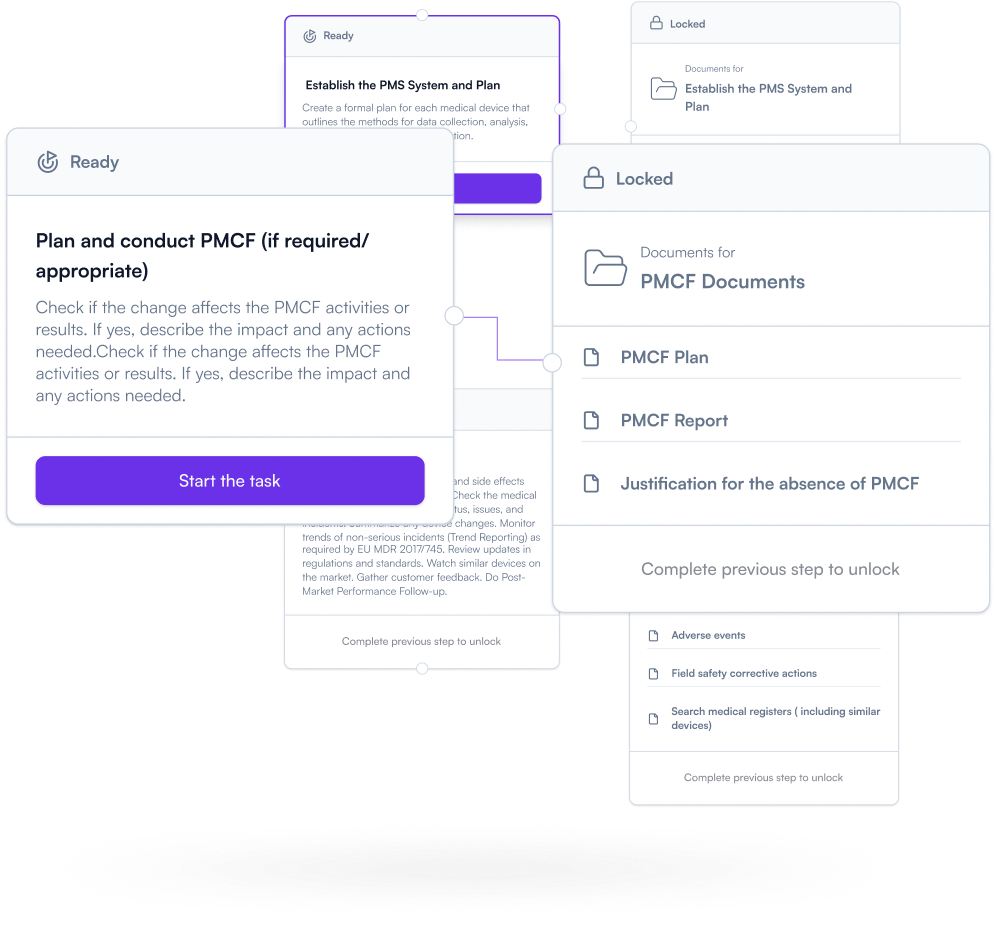
Don't treat PMS (Post-market surveillance) as an afterthought. Plan your post-market activities early and keep documentation aligned as real-world feedback and changes come in. Keep your PMS evidence audit-ready — continuously.
News and insights on health tech regulation, MDR compliance, and medical device pathways.

In my last post, I shared why we built Health Tech Pathways. This one is about how we designed it — a look into our engine room. But how did we end up with this design and user experience? It started with the customers We have seen the frustration in the eyes of start-up founders when they realise how difficult, expensive, and time-consuming regulatory approval is. After we received the grant of EUR 4.2m from the Novo Nordisk Foundation in October 2024, we decided to understand that frustra

I'm often asked why we built Health Tech Pathways. So here it is. It started on a dockside In 2018, I gathered the first seven health tech start-ups on a dockside in Copenhagen. That was the beginning of Health Tech Hub Copenhagen. Since then, we have supported more than 120 start-ups and scale-ups, reaching more than 5 million people with innovative solutions. Along the way, one pattern kept repeating. Every start-up faces the same three barriers: 1. Funding 2. Market access — (i.e. f
Disrupting Healthcare wrote this long article about the challenges with EU MDR and Health Tech Pathways - in English here: Health Tech Pathways and the Bottleneck Upstream of EU MDR - disrupting.healthcare
Clear answers for teams building regulated products.
Still have questions?
Book a demo with one of our team members.
Whether you're just getting started or already building, get clarity on your regulatory path and access the full workflow with Health Tech Pathways.